Research
Permeation of small-molecule drugs through the lipid bilayer
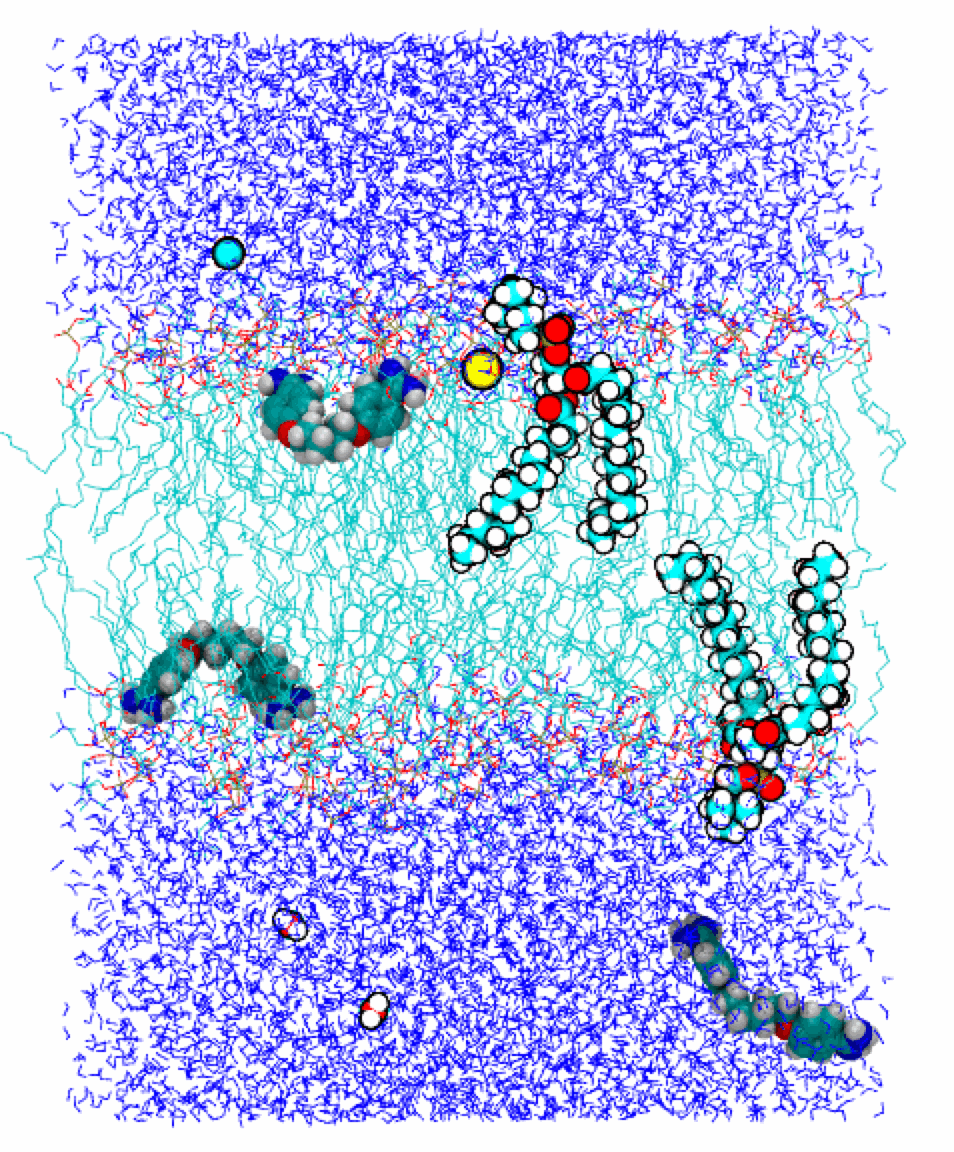
The cell membrane separates the inside of a cell from its outside by preventing many small molecules from freely entering or leaving the cell. This task is mainly carried out by the lipid component of the membrane, which is a self-assembled double layer of lipid molecules (two of them shown in the figure on the left). The lipid bilayer consists of a hydrophobic layer (cyan) sandwiched by two hydrophilic layers. The latter are immersed in the surrounding water (dark blue).
Most drug molecules of relatively small molecular weight permeate the cell membrane by crossing the lipid bilayer without the help of protein channels or transporters. In other words, they face the challenge of crossing a layered heterogeneous environment. We investigate the mechanisms allowing some of these drug molecules to traverse the lipid bilayer. Performing molecular dynamics simulations we quantify the energetics and dynamics of the permeation process.
Related references: 1. Oruç, Küçük, Sezer, Lipid bilayer permeation of aliphatic amine and carboxylic acid drugs: Rates of insertion, translocation and dissociation from MD simulations, PCCP, 18: 24511 (2016). 2. Sezer, Oruç, Protonation kinetics compromise liposomal fluorescence assay of membrane permeation, JPC B, 121: 5218 (2017).
Funding: TÜBA-GEBİP (Turkish Academy of Sciences Outstanding Young Scientist) Award (2014-2017).
Solvent dynamics and dynamic nuclear polarization (DNP)
To smoothly perform their biological functions proteins and nucleic acids need to be constantly lubricated. During the evolution of life water has established itself as an omnipotent lubricant of biomacromolecules. The water molecules in the immediate vicinity of a biomolecule are, therefore, expected to play a special role in facilitating its functional motions. Such waters, forming the so-called “hydration layer” of the macromolecule, are expected to differ from waters in the bulk. However, quantitative measurements of the static and dynamic properties of solvent molecules in the hydration layer are hard to obtain. In this regard, dynamic nuclear polarization is a very promising technique for quantifying how solvent molecules at protein-water or lipid-water interfaces move around.
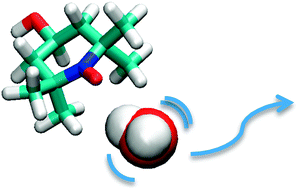 Performing molecular dynamics simulations of polarizing agents in water and other solvents, we calculate the magnitude of the DNP effect that would be measured if the solvent motions in the computer corresponded to the real-world motions. We then compare with actual experimental data to both validate our simulations and to provide a detailed explanation of the molecular processes behind the measurements.
Performing molecular dynamics simulations of polarizing agents in water and other solvents, we calculate the magnitude of the DNP effect that would be measured if the solvent motions in the computer corresponded to the real-world motions. We then compare with actual experimental data to both validate our simulations and to provide a detailed explanation of the molecular processes behind the measurements.
Related references: 1. Sezer, Computation of DNP coupling factors of a nitroxide radical in toluene: Seamless combination of MD simulations and analytical calculations, PCCP, 15:526 (2013). 2. Küçük, Neugebauer, Prisner, Sezer, Molecular simulations for dynamic nuclear polarization in liquids: A case study of TEMPOL in acetone and DMSO, PCCP, 17:6618 (2015). 3. Küçük, Sezer, Multiscale computational modeling of 13C DNP in liquids, PCCP, 18:9353 (2016).
Funding: TÜBİTAK 3501 Career Grant “Calculation of dynamic nuclear polarization transfer in liquids through multi-scale modeling of molecular motions and spin interactions”. Grant number: 112T770 (2013-2015).
Dynamic nuclear polarization at the lipid bilayer interface
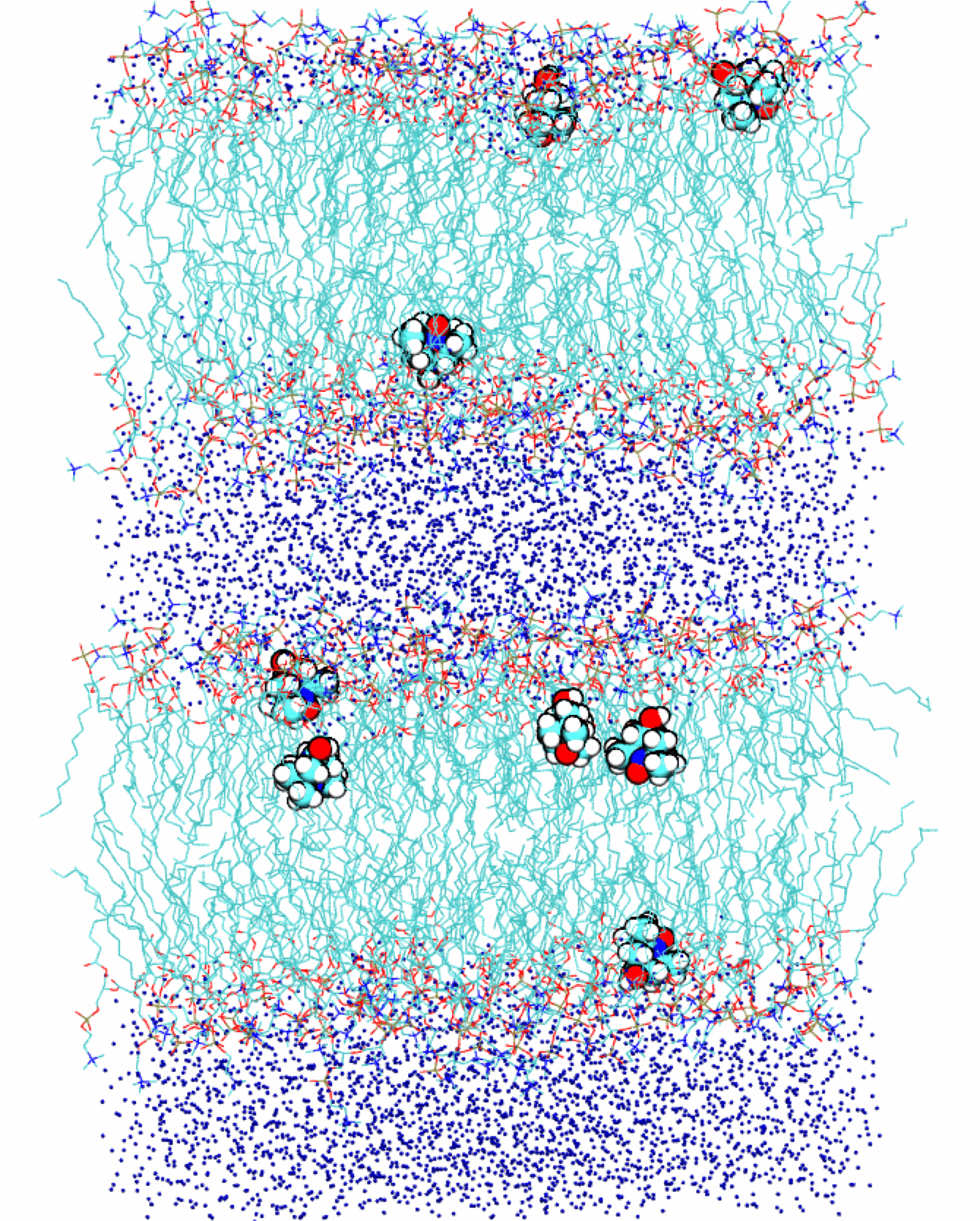 Recently it has been possible to increase the nuclear magnetic resonance (NMR) signal from lipids in lipid bilayers employing dynamic nuclear polarization at 9.2 Tesla. It is not clear, however, what molecular motions contribute to the effect. To elucidate the nature of these motions we perform molecular dynamics simulations of the polarizing molecules in the heterogeneous environment of stacked lipid bilayers (as shown on the right).
Recently it has been possible to increase the nuclear magnetic resonance (NMR) signal from lipids in lipid bilayers employing dynamic nuclear polarization at 9.2 Tesla. It is not clear, however, what molecular motions contribute to the effect. To elucidate the nature of these motions we perform molecular dynamics simulations of the polarizing molecules in the heterogeneous environment of stacked lipid bilayers (as shown on the right).
Another experimental possibility is the polarization of water protons due to spin-labeled lipids that are part of the lipid bilayer. Such measurements aim to quantify the local water diffusivity both at the polar surface and in the hydrophobic interior of the lipid bilayer. Because atomic polarizability is expected to be essential for the realistic modeling of the water dynamics at interfaces, we will perform MD simulations of lipid bilayers with spin-labeled lipids using a polarizable molecular force field.
Related references: 1. Jakdetchai, Denysenkov, Becker-Baldus, Dutagaci, Prisner and Glaubitz, Dynamic nuclear polarization-enhanced NMR of aligned lipid bilayers at ambient temperature, JACS, 136:15533 (2014). 2. Kausik, Han, Dynamics and state of lipid bilayer-internal water unraveled with solution state 1H dynamic nuclear polarization, PCCP, 13:7732 (2011). 3. Song, Franck, Pincus, Kim, Han, Specific ions modulate diffusion dynamics of hydration water on lipid membrane surfaces, JACS, 136:2642 (2014).
Biomolecular dynamics and electron spin resonance (ESR) spectroscopy
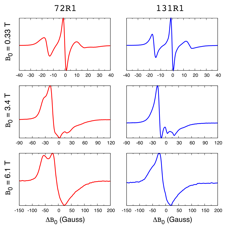 ESR spectroscopy is extensively used to probe the dynamics of biomolecules ranging from proteins, to nucleic acids, and lipids. In this technique, a spin label is covalently attached to the biomolecule of interest. The figure on the left, for example, shows the ESR spectra of the protein T4 Lysozyme recorded at three different magnetic fields. The red spectra correspond to a spin label attached to amino acid at position 72 in the sequence of T4 Lysozyme. The blue spectra correspond to a spin label attached at position 131. The differences in the red and blue ESR spectra indicate that the local environment and the dynamics of the spin labels at these two positions are different. What exactly those differences are and how they relate to the structure and dynamics of the protein is hard to say by looking at the spectra. We perform molecular dynamics (MD) simulations of spin-labeled proteins and DNA to unravel the molecular processes behind such ESR spectra in atomic detail.
ESR spectroscopy is extensively used to probe the dynamics of biomolecules ranging from proteins, to nucleic acids, and lipids. In this technique, a spin label is covalently attached to the biomolecule of interest. The figure on the left, for example, shows the ESR spectra of the protein T4 Lysozyme recorded at three different magnetic fields. The red spectra correspond to a spin label attached to amino acid at position 72 in the sequence of T4 Lysozyme. The blue spectra correspond to a spin label attached at position 131. The differences in the red and blue ESR spectra indicate that the local environment and the dynamics of the spin labels at these two positions are different. What exactly those differences are and how they relate to the structure and dynamics of the protein is hard to say by looking at the spectra. We perform molecular dynamics (MD) simulations of spin-labeled proteins and DNA to unravel the molecular processes behind such ESR spectra in atomic detail.
Related references: 1. Sezer, Freed, Roux, Multifrequency electron spin resonance spectra of a spin-labeled protein calculated from molecular dynamics simulations, JACS, 131:2597 (2009). 2. Sezer, Sigurdsson, Simulating electron spin resonance spectra of macromolecules labeled with two dipolar-coupled nitroxide spin labels from trajectories, PCCP, 13:12785 (2011). 3. Halbmair, Seikowski, Tkach, Höbartner, Sezer, Bennati, High-resolution measurement of long-range distances in RNA: pulse EPR spectroscopy with TEMPO-labeled nucleotides, Chem Sci, 7:3172 (2016).
Funding: TÜBİTAK 1001 Research Grant, “Analysis of DNA structure and motions in the presence of mismatch using MD simulations and calculation of ESR spectra”. Grant number: 111T961 (2012-2014).